
Actas del Congreso Nacional de
Tecnología Aplicada a Ciencias
de la Salud

Actas del Congreso Nacional de Tecnología Aplicada a Ciencias de la Salud Vol. 4, 2022
La transformación del melanocito a melanoma cutáneo (MC) y hasta melanoma metastásico, involucra la regulación epigenética ejercida por microRNAs (miRNAs). El objetivo, fue establecer miRNAs como potenciales biomarcadores de la progresión en MC, comparando 19 estudios que reportaban expresión diferencial de microRNAs en muestras de pacientes con MC en diferentes estados de progresión y 43 estudios con confirmación experimental de la unión entre miRNAs y sus mRNAs blanco. Se observaron coincidencias para los miR-211-5p, miR-203, miR-137 y miR-205 a la baja; y para los miR-21, miR-17, miR-106a, miR-93, miR-25 y miR-92 al alta. El enriquecimiento mediante vías KEGG, reveló la potencial regulación de estos microRNAs sobre programas de proliferación, crecimiento y migración, entre otros.
Palabras Claves: miRNAs, Progresión Tumoral, Melanoma
Melanocyte transformation to cutaneous melanoma (CM) and even metastatic melanoma involves epigenetic regulation by microRNAs (miRNAs). The objective was to establish miRNAs as potential biomarkers of progression in CM, comparing 19 studies reporting differential expression of miRNAs in samples from patients with CM at different stages of progression and 43 studies with experimental confirmation of binding between miRNAs and their target mRNAs. Matches were observed for miR-211-5p, miR-203, miR-137 y miR-205 at low; and for miR-21, miR-17, miR-106a, miR-93, miR-25, and miR-92 at high. Enrichment by KEGG pathways revealed the potential regulation of these microRNAs on proliferation, growth, and migration programs, among others.
Key Words: miRNAs, Tumor Progression, Melanoma
Cáncer es el nombre común para referirse a un conjunto de más de 100 enfermedades neoplásicas, en las cuales un tipo celular desarrolla una serie de huellas que abarcan mantenimiento de la señalización proliferativa, desregulación del metabolismo celular, evasión de supresores de crecimiento, evasión del sistema inmune, habilitación de la inmortalidad replicativa, evasión de muerte celular, inducción de angiogénesis y activación de invasión y metástasis [1]. Este proceso multietapas involucra alteraciones genéticas y epigenéticas que de manera acumulada y en conjunto, podrían conferir ventaja selectiva a algunas células tumorales y generar una alta heterogeneidad celular y molecular, condiciones que podrían relacionarse con la progresión tumoral.
El MC, es un cáncer de melanocitos, células especializadas encargadas de la síntesis de melanina, es altamente agresivo y metastásico, de diagnóstico tardío y bajo pronostico en etapas avanzadas de progresión y aunque representa tan sólo el 4% de los tumores malignos de piel, es el responsable del 80% de la mortalidad por este cáncer en el mundo [2].
El proceso multietapas que conduce la transformación del melanocito hasta melanoma metastásico, conocido como progresión tumoral, se ha caracterizado histopatológicamente en tres estados de progresión: nevus displásico, melanoma primario de crecimiento en fase radial (RGP) que puede conducir a un melanoma de crecimiento en fase vertical (VGP) y finalmente melanoma metastásico [3]. El grado de avance del MC se correlaciona con bajo pronóstico y sobrevida; a la fecha no existe tratamiento efectivo para estados avanzados de la enfermedad. Los mecanismos celulares y moleculares asociados son un foco actual de investigación; varios cambios sobre oncogenes y genes supresores tumorales asociados a alteraciones genéticas y epigenéticas han sido reportados.
Los miRNAs son pequeños RNAs, no codificantes reguladores postranscripcionales de la expresión de al menos el 60% de los genes que codifican proteínas en mamíferos de una amplia variedad de procesos celulares [4]. Variaciones en la expresión de los miRNAs podrían asociarse con la regulación de diferentes programas celulares alterados y descritos como huellas del cáncer antes citados, en la progresión del melanoma. El establecimiento de miRNAs específicos durante cada etapa de la progresión del melanoma permitiría proponer potenciales marcadores, así como establecer nuevas estrategias de diagnóstico, tratamiento y prevención para esta enfermedad.
Con el propósito de asociar la regulación que ejercen los miRNAs sobre sus mRNAs blanco y la progresión en melanoma cutáneo; en este documento, se describen mediante ejemplos específicos encontrados en la literatura científica, algunos cambios en la expresión de grupos de miRNAs en diferentes estados de la progresión y su asociación con la regulación de procesos celulares.
2.1 Melanoma cutáneo y su progresión
El MC representa el 1% de los cánceres de piel, pero es el responsable de más del 80% de las muertes por este tipo de cáncer. Para el 2020 se diagnosticaron 325.000 casos nuevos de melanoma, lo que representa el 1.7% de los diagnósticos globales de cáncer, con una incidencia en hombres de 3,8 y en mujeres de 3 por cada 100.000 habitantes [5].
Para describir la progresión del MC, se cuenta con un sistema de grados de progresión del 0 al IV propuesto por el Comité Conjunto Americano contra el Cáncer (AJCC) que tiene como criterios principales el tamaño de la masa tumoral, su ubicación y la posibilidad de invasión a otros órganos. Esta caracterización, se complementa con el sistema (TNM), T para describir el espesor del tumor primario; N, para informar presencia de células cancerosas en ganglios linfáticos y M para describir metástasis [6]. El grado de progresión del MC presenta una correlación inversa con el pronóstico de la enfermedad, en estadio I, hasta 90% de supervivencia, que se reduce a un 60 % en estadio II, a un 10 % para el estadio III y con mortalidad total en estadio IV [7].
Para avanzar en el conocimiento de los determinantes moleculares de la progresión del MC, se han realizado aproximaciones omicas que comparan datos experimentales en muestras de tejidos de pacientes diagnosticados con melanoma en diferentes estadios de progresión o líneas celulares derivadas de melanoma, con muestras de tejidos no tumorales o líneas derivadas de melanocitos [8]. Cerca del 10% de los casos de melanoma familiar, involucran mutaciones puntuales en el gen supresor tumoral p16INK4a (CDKN2A) y alteraciones en las vías MAP-quinasa, PI3K/AKT, WNT y MITF, así como mutaciones de ganancia de función en los oncogenes BRAF, NRAS, KIT entre otros [9].
Recientemente, se ha propuesto correlacionar un modelo de progresión del MC con la adquisición de cambios genéticos y epigenéticos. El modelo, presenta una secuencia común de eventos que conduce a la transformación del melanocito hasta melanoma metastásico, donde cada etapa sugerida adiciona cambios con respecto a su predecesora en función de ganancia de mutaciones específicas sobre reguladores mitogénicos como por ejemplo BRAF, NRAS, MYC, en nevus benigno; supresores de la senescencia, como la deleción de CDKN2A en nevus displásico y melanoma RGP; mutaciones de genes que participan en apoptosis en melanoma VGP y mutaciones asociadas con la inmortalización como la expresión de TERT. Sin embargo, se ha observado que no todos los melanomas presentan los mismos cambios, ni el mismo orden de eventos a través de cada uno de los tipos de lesión, por lo que se hace necesario explorar otros mecanismos y moléculas asociadas con la regulación de la expresión génica y la progresión tumoral en MC que no sean explicados solamente a partir de variaciones en la secuencia de DNA.
2.2 miRNAs y progresión tumoral en melanoma cutáneo.
Del total de casos de MC reportados, el 10% estaría asociado a melanoma familiar y solo el 40% de los casos esporádicos presenta alteraciones en oncogénes[10]. Otras aproximaciones se enfocan en el estudio de las variaciones epigenéticas como silenciamiento de genes supresores tumorales mediante la metilación del DNA en regiones promotoras, activación de oncogenes, remodelación de la cromatina y regulación mediada por RNAs no codificantes, como los miRNAs; estos últimos, a través de su regulación de genes implicados en diferentes procesos celulares como proliferación, muerte, control de ciclo celular, senescencia, angiogénesis, diferenciación e incluso metástasis[11].
Los miRNAs son RNAs pequeños no codificantes, entre 18-25 nt de longitud, la mayoría de los miRNAs se derivan de RNAs largos de cadena doble intramolecular, que se escinden secuencialmente por acción de RNasas tipo III, primero en el núcleo y después en el citoplasma, hasta generar un duplex de microRNA. Tras etapas posteriores, una de las hebras del duplex se asocia a un complejo de silenciamiento inducido por RNA (miRISC) que desencadena la disminución en un mRNA específico por degradación del transcrito o una represión de la traducción del mRNA a proteína [12]. La probabilidad de complementariedad entre la secuencia de un microRNA y el 3´UTR del RNA codificante diana o blanco puede aumentar como consecuencia de los alineamientos múltiples entre varios miRNAs y diferentes regiones 3´UTR de un mismo mRNA, o de los alineamientos entre los 3´UTR de diferentes mRNAs y la secuencia de un mismo microRNA; lo que sugiere que un mismo miRNA tendría la posibilidad de regular diferentes mRNAs, y a su vez, un mismo mRNA puede ser regulado por diferentes miRNAs [13].
Varios estudios se han focalizado en evaluar los niveles de expresión de subconjuntos de miRNAs, a través del análisis completo del perfil de expresión obtenido por diferentes métodos como microarreglos, ensayos de RT-qPCR, y secuenciación de última generación (NGS) y su asociación funcional con blancos mRNAs específicos en la evaluación de programas celulares en melanoma [14]. No obstante, la evidencia experimental es limitada, en parte, debido a los modelos implementados que se fundamentan generalmente, en análisis de expresión y cuantificación de miRNAs desde muestras independientes de pacientes categorizados histopatológicamente en estadios discretos de la progresión y no desde muestras procedentes del mismo tejido a través de la progresión. La alta variabilidad asociada a las muestras (cultivos primarios, líneas celulares), la cantidad de datos a analizar y las condiciones experimentales diferenciales asociadas al uso de diferentes técnicas y plataformas (microarreglos-qRT-PCR-NGS), así como su validación funcional dificultan identificar los microRNAs alterados en MMC durante su progresión [14].
3.1 Tipo de estudio
Revisión sistemática de la literatura con enfoque cualitativo.
Se seleccionaron 19 estudios donde se comparaba la expresión de miRNAs en muestras de tejidos de pacientes diagnosticados con melanoma en diferentes estados de progresión, incluyendo melanocitos, nevus común y líneas celulares derivadas de melanoma. Se revisaron 43 estudios adicionales donde se evaluó la asociación funcional entre un miRNA diferencialmente expresado y su mRNA diana. Se establecieron 5 categorías que intentan recoger los diferentes estados de la progresión en melanoma cutáneo: de melanocito a melanoma primario, de nevus común a melanoma primario, de melanocito a melanoma metastásico, de nevus a melanoma metastásico y de melanoma primario a melanoma metastásico. En las figuras 1 y 2 se relaciona en detalle el diseño metodológico, así como los criterios de elegibilidad.
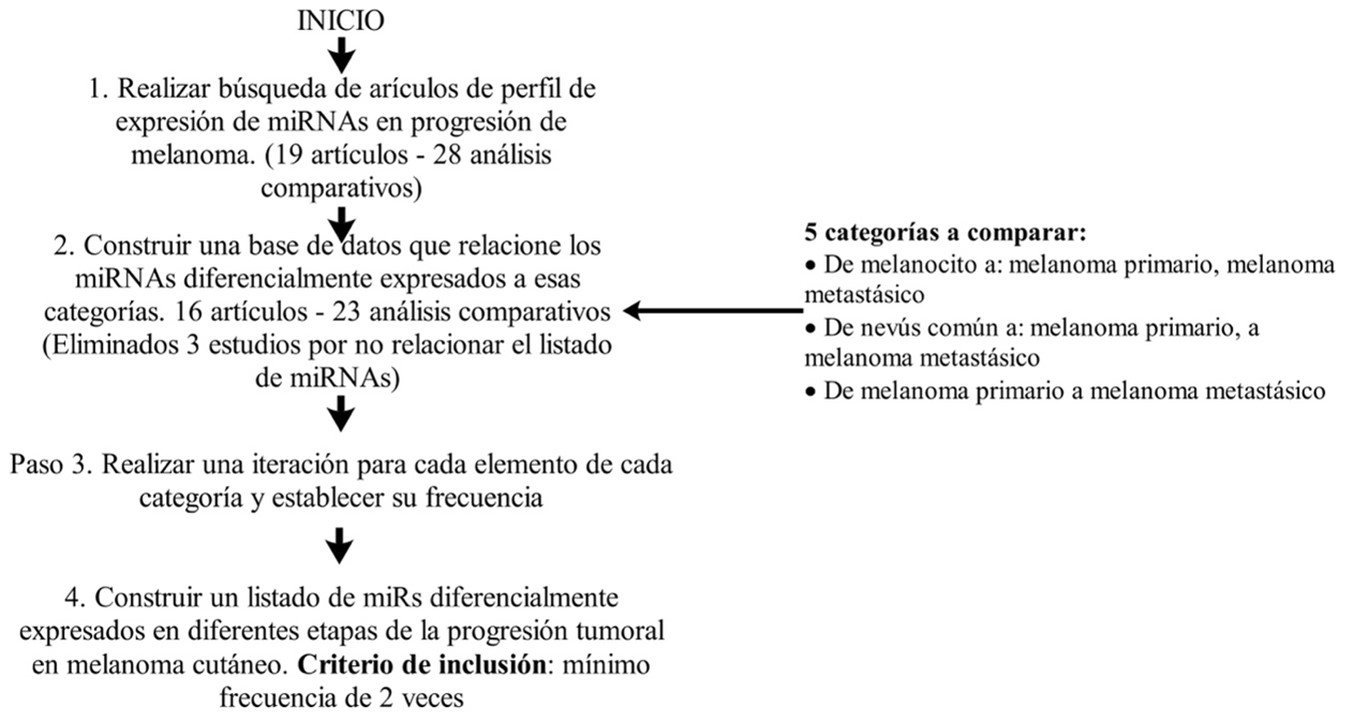
Figura 1. Resumen metodológico para establecer un perfil de miRNAs diferencialmente expresados en MC
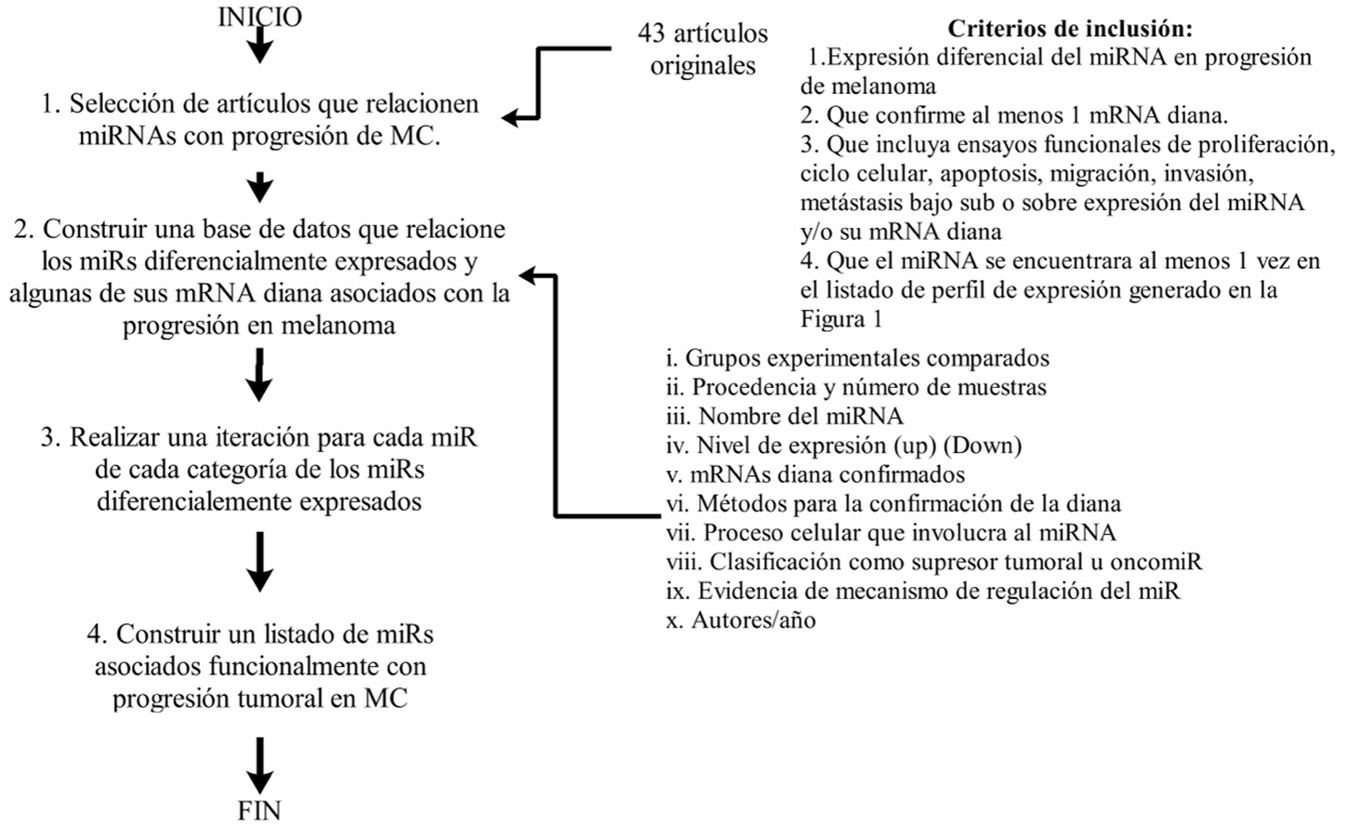
Figura 2. Resumen metodológico para establecer miRNAs diferencialmente expresados con mRNAs confirmadas experimentalmente
Considerando las limitaciones antes descritas sobre el análisis de miRNAs individuales para explicar fenómenos complejos de regulación de expresión génica durante la progresión de MC, se han propuesto otras aproximaciones como el análisis comparativo entre los perfiles de expresión diferencial de mRNAs y de miRNAs en una misma muestra, donde los análisis de correlación inversa, sumados al establecimiento de los blancos del miRNA y su enriquecimiento funcional, facilitan las tareas de confirmación experimental. En la tabla 1 se relaciona un listado de miRNAs diferencialmente expresados a través de estadios discretos de la progresión tumoral en MC.
Tabla 1. Listado de miRNAs diferencialmente expresados y con confirmación experimental sobre mRNAs blanco asociados con la progresión del melanoma cutáneo
| Etapa discreta de progresión | Melanoma primario con respecto a melanocito | Melanoma primario con respecto a nevus común | Melanoma metastásico con respecto a melanocito | Melanoma metastásico con respecto a nevus común | Melanoma metastásico con respecto a melanoma primario |
|---|---|---|---|---|---|
| Expresión de miRNAs a la baja | miR-26b, miR-125b, miR-137, miR-211, miR-664, miR-135a, miR-769 | let-7b, miR-18b, miR-1280 | miR-33a, miR-203, miR-524-5p, miR-573, miR-137, miR-125b, miR-382, miR-143, miR-31, miR-200c, miR-205, let-7i, miR-34, miR-34a, miR-30b, miR-22, miR-138, miR-451a-1, let-7a | miR-193b, miR-205 | miR-34b, miR-148, miR-137 |
| Expresión de miRNAs al alta | miR-135a, miR-769 | miR-506-514, miR-340, miR-214, miR-17, miR-106a, miR-93, miR-25, miR-92 | miR-638, miR-21, miR-182, miR-125b |
Sobre los miRNAs 211, 137 y 125b se ha reportado su asociación en MC. El miR-211 se expresa diferencialmente entre los melanocitos normales y líneas celulares de melanoma no pigmentadas y los melanomas primarios de pacientes, disminuyendo el crecimiento y la invasión celular [4]. En cuanto a pigmentación el miRNA 211-5p ha sido propuesto como un regulador indirecto de la pigmentación a través de la unión al receptor TGF β-2, un factor responsable de la transcripción de los genes asociados a la pigmentación Dct, TYRP1, TYR y PMEL17 y la unión al mRNA del inhibidor de TYR, EDEM1; además, MITF puede regular la expresión de miR-211-5p [15]
El miR 125b-3p asocia con regulación de la melanogénesis en estado estacionario. La expresión de miR-125b es inversamente proporcional a los niveles de pigmento y su expresión está regulada por la concentración de cAMP [16]. De hecho, en eventos invasivos y metastásicos, los miR-221 y miR-222, que provienen de un precursor común, se encuentran sobre expresados en fenotipos de melanoma más invasivos, disminuyen la expresión de su blanco molecular común c-Kit y aumentan la proliferación acelerando en ciclo celular [17]. Entre tanto, la familia let-7 se encontró desregulada en melanoma cutáneo maligno en comparación con los nevus benignos, algunos de los blancos moleculares de los miRNAs pertenecientes a esta familia son NRAS, MYC, CCND1, CCND3, CDK4 e integrina β3, relacionados con proliferación, migración e invasión [18]. El miR-125b se une a AKT3, BCL2 y E2F2 influenciando la sobrevivencia celular e induciendo senescencia en melanomas humanos [19]. Respecto a proliferación celular, se han estudiado los miR-137, miR-148 y miR-182 [20] que se encuentran sub expresados en melanoma, son reguladores negativos de señales proliferativas mediante la unión a MITF.
El estudio actual de perfiles de expresión de cientos de microRNAs ha permitido plantear efectos probablemente sinérgicos de control postranscripcional, en donde conjuntos de miRs se expresan para desarrollar o mantener un programa celular, una hipótesis que aún no se ha comprobado, en parte por el desconocimiento de los mecanismos que regulan la expresión de los miRs y de sus diferentes niveles de expresión, que en conjunto sugieren una maquinaria molecular que podría operar en redes de regulación miRs-mRNAs-miRs.
Se generaron en la plataforma miRNet, 5 redes de regulación entre miRNAs, factores de transcripción y genes por estadio discreto de progresión, ver tabla 2. En la Figura 3, se presenta el análisis de enriquecimiento funcional por vías de señalización KEGG para los miRNAs descritos en la Tabla 1.
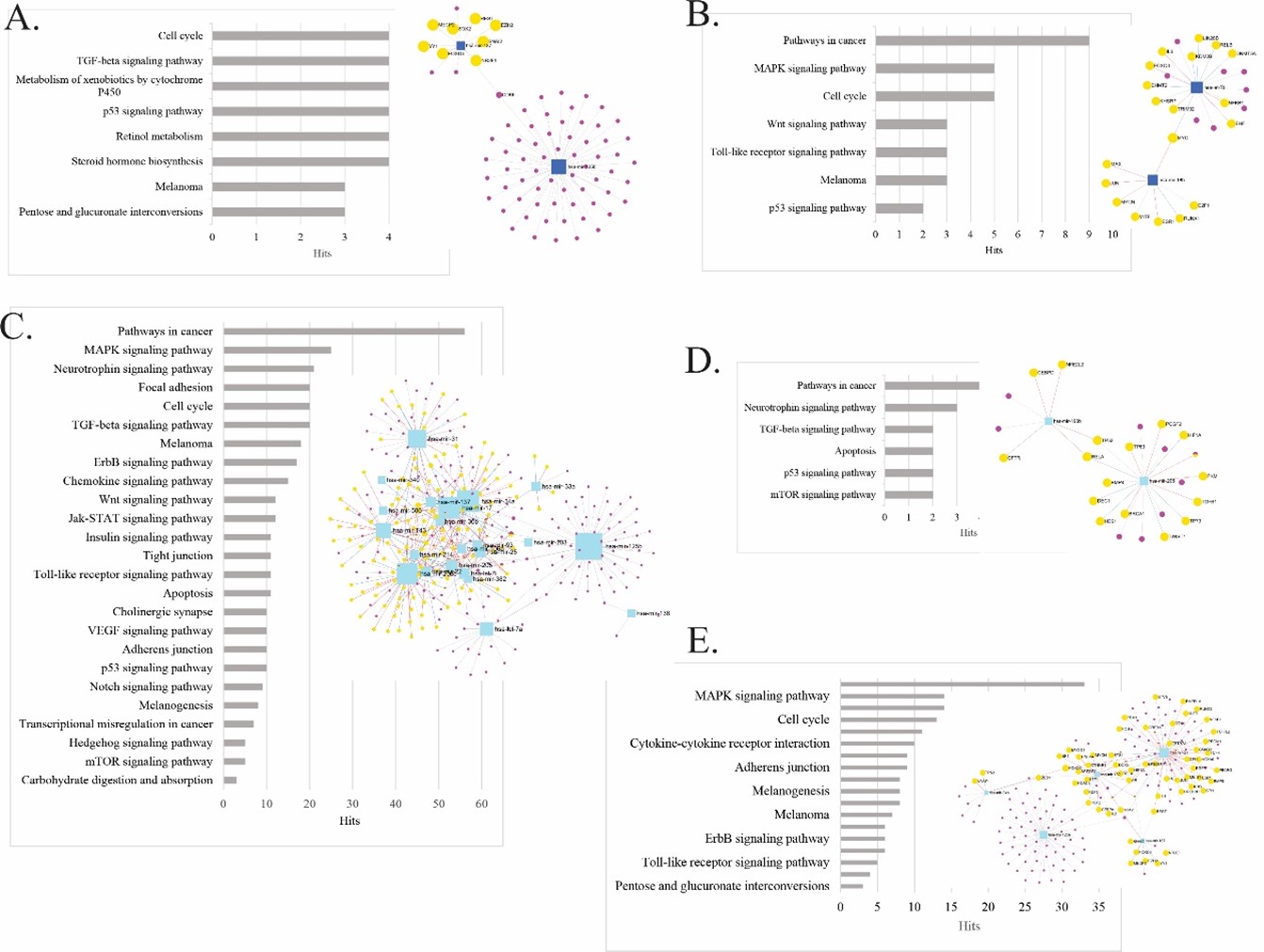
Figura 3. Análisis de enriquecimiento funcional por vías de señalización KEGG de las potenciales interacciones entre los miRNAs diferencialmente expresados y sus blancos moleculares en un modelo de red
Tabla 2. Información sobre las redes generadas en miRNet de regulación entre miRNAs, factores de transcripción y genes asociados con la progresión del melanoma cutáneo
| Etapa discreta de progresión | Melanoma primario con respecto a melanocito | Melanoma primario con respecto a nevus común | Melanoma metastásico con respecto a melanocito | Melanoma metastásico con respecto a nevus común | Melanoma metastásico con respecto a melanoma primario |
|---|---|---|---|---|---|
| Factores de transcripción - TF | 8 | 19 | 143 | 17 | 65 |
| Genes - mRNAs blanco | 67 | 8 | 188 | 9 | 124 |
| miRNAs confirmados experimentalmente | 2 | 2 | 22 | 2 | 5 |
| Topología de la red - número de puentes | 76 | 28 | 462 | 27 | 199 |
La tendencia en la investigación en miRNAs y MC ha evolucionado de asociar funcionalmente miRNAs específicos alterados a explorar redes moleculares y conexiones entre miRs y redes de genes, vías de señalización, microambiente y otros mecanismos de regulación durante las etapas de la progresión tumoral. Se espera que esta aproximación facilite el planteamiento de modelos hipotéticos de la evolución y progresión tumoral centrados en miRNAs, que podrían integrar cambios genéticos, epigenéticos, histopatológicos, clínicos y biológicos, lo cual tendría un impacto en potenciales nuevos biomarcadores y por ende en las estrategias futuras de prevención, diagnóstico y tratamiento del MC.