
Actas del Congreso Nacional de
Tecnología Aplicada a Ciencias
de la Salud

Actas del Congreso Nacional de Tecnología Aplicada a Ciencias de la Salud Vol. 5, 2023
La Enfermedad de Creutzfeldt-Jakob es una enfermedad rara causada por la proteína prión, caracterizada por demencia rápidamente progresiva. Actualmente no se tiene cura y la mayoría de los pacientes fallecen tras un año de presentar síntomas. En este estudio se busca la concepción y creación de la primera base de datos en México orientada hacia aquellos individuos que han sido afectados por este padecimiento. Para alcanzar esta meta, se adoptó la metodología PRISMA, la cual rigurosamente establece la búsqueda y selección de casos de pacientes mexicanos, consolidando esta información con los registros extraídos del Instituto Nacional de Ciencias Médicas y Nutrición Salvador Zubirán y del Instituto Nacional de Neurología y Neurocirugía, con ello se construyó el primer banco de 74 pacientes. Se aplican estimaciones no paramétricas de Kaplan-Meier las cuales arrojan que pacientes de México de alrededor de 20 años tienen una supervivencia media mayor a un año, superando sus homólogos de otras poblaciones. Se hipotetiza al factor ascendente Nativo Americano como protector de nuestra población.
Palabras claves: Prion diseases, Creutzfeldt-jakob-syndrome; Mexico
Creutzfeldt-Jakob disease is a rare disease caused by the prion protein, characterized by rapidly progressive dementia. Currently there is no cure and most patients die within a year of presenting symptoms. The main objective of this study is the conception and creation of the first database in Mexico specifically oriented toward those individuals who have been affected by this disease. To achieve this goal, the PRISMA methodology was adopted, which rigorously establishes the search and selection of cases of Mexican patients, consolidating this information with the records extracted from the Instituto Nacional de Ciencias Médicas y Nutrición Salvador Zubirán and the Instituto Nacional de Neurología y Neurocirugía, thus building the first bank of 74 patients. Non-parametric Kaplan-Meier estimates are applied, which show that patients from Mexico aged around 20 years have a mean survival of more than one year, surpassing their counterparts from other populations. Native American ancestry is hypothesized as a protective factor in our population.
Keywords: Prion diseases, Creutzfeldt-jakob-syndrome; Mexico
Cuando nos enfermamos, por lo regular se busca la asociación hacia agentes causales como virus, bacterias, parásitos u hongos, sin embargo, actualmente se puede añadir una nueva clasificación de agentes causales: Los priones. El nacimiento del término surge desde 1972, cuando el Dr. Stanley B Prusiner durante su estancia como residente de neurología atiende a un paciente que presenta Enfermedad de Creutzfeldt-Jakob, lo cual en aquella época era asociado a un “virus de lento crecimiento”, aún no existía el término prión [1]. En 1997, gracias a las investigaciones del Dr. Prusiner, le es otorgado el Premio Nobel en Fisiología o Medicina por “el descubrimiento de los Priones - un nuevo principio biológico de infección”, con ello se acuña el nuevo término como el organismo causante de enfermedades [2].
Los priones son catalogados como agentes proteináceos mal plegados, causantes de encefalopatías espongiformes transmisibles que afectan de forma mortal al portador, el cual puede ser tanto humano como animal [3]. Las Enfermedades Priónicas agrupan una variedad de presentaciones clínicas como lo son: Enfermedad de Creutzfeldt-Jakob (ECJ), Insomnio Familiar Fatal (IFF), Kuru, Enfermedad de Huntington, Prionopatía sensible a proteasa y Gerstmann-Sträusller-Scheinker [4]; la incidencia se asocia en un caso por millón de habitantes, sin embargo, la descripción epidemiológica a nivel mundial está mejor detallada en países ubicados en América del Norte (exceptuando México), Europa, Japón y Australia, dejando sin datos a naciones emergentes [3]. La preocupación hacia este tipo de patología reside en que los priones requieren un tiempo de incubación de hasta 40 años en el portador (temporada durante la cual no se presentarán ni signos ni síntomas), y tras la presentación de los primeros síntomas, la supervivencia media es de apenas un año [3], aunado a que, hasta el momento, no existe tratamiento alguno para los pacientes, solamente se utilizan algunos fármacos con efecto paliativo [5].
2.1 Prión Homeostático (PrPc) vs Prión Patológico (PrPSc)
Los priones se encuentran de manera natural en humanos y animales, específicamente a nivel cerebral, mucho se especula aún sobre su función principal, sin embargo, se teoriza sobre su posible participación en el transporte de metales pesados, específicamente de Cu2+ y Zn2+ [6]. La presencia de PrPSc no incentiva el desarrollo de la respuesta inmune efectiva, en cambio, eslabones del sistema inmunológico, como los ganglios linfáticos, sirven como sitio de incubación y replicación del PrPSc. No se ha detectado función principal del PrPSc, más que la propiedad de replicación en semilla y conversión del PrPc. El principal cambio ocurre a nivel molecular, siendo el PrPSc rico en láminas Beta, mientras que el PrPc teniendo en su mayoría Alpha-Hélices [7].
2.2 Clasificación Etiológica
Los priones pueden transmitirse entre animales y humanos, a través de vía horizontal [8], en el caso de animales se tiene registro de la “Enfermedad de Vacas Locas”, la cual azotó al ganado de Francia e Inglaterra el siglo pasado. Para los humanos, la ECJ puede presentarse de manera esporádica, siendo la forma más común en los pacientes, reportando asociación hasta de un 85% de los casos [9], no se conoce detalladamente el motivo del cambio estructural de un PrPc hacia un PrPSc cuando se realiza de forma esporádica. Un 15% de los casos se asocian por mutación genética, siendo la segunda causa más común de ECJ, la anomalía ocurre en el gen PRNP, encargado de la codificación de la proteína prión, la cual de manera homeostática se encarga del transporte de metales pesados [10]; la presentación Familiar sigue una herencia mendeliana autosómica dominante en la mayoría de los casos [9]. Otra vía de infección de priones patológicos es la vía adquirida [9], menos común, la cual es a través de procedimientos quirúrgicos que usen órganos inmunoprivilegiados como sitio de acceso, o a través de la ingesta de alimentos infectados con priones.
2.3 Criterios de Diagnóstico
El diagnóstico de ECJ se divide en 3 tipos: Definitivo, Probable y Posible (ver Tabla 1); esto depende directamente de la cantidad de estudios de laboratorio y gabinete disponibles para la confirmación y de la fase de presentación del paciente. El problema en este punto radica en que, gran cantidad de los casos reportados para México manifiestan pacientes con ECJ con diagnóstico Posible, lo cual, puede ser en realidad Alzheimer, Parkinson, o alguna otra Demencia Rápidamente Progresiva no Priónica.
Tabla 1. Criterios de Diagnóstico para ECJ
| Signos y Síntomas | |
|---|---|
| I | A. Demencia Rápidamente Progresiva |
| II | A. Mutismo Acinético B. Mioclonías C. Signos Piramidales o Extrapiramidales D. Alteraciones Visuales o Cerebelares |
| Clasificación | |
| Posible | IA + ≥2 síntomas de II + Duración <2 años |
| Probable | IA + ≥2 síntomas de II + EEG característico*; o IA + ≥2 síntomas de II + MRI característico*; o IA + ≥2 síntomas de II + Prueba de Proteína 14-3-3 [Positiva]; o Síndrome Neuropsiquiátrico Progresivo + Prueba RT-QUIC [Positiva] |
| Definitivo | Síndrome Neuropsiquiátrico Progresivo + Confirmación Neuropatológica; o Confirmación Inmunocitoquímica; o Confirmación Bioquímica |
| *Ver Sección 2.4 Biomarcadores y Estudios de Gabinete | |
2.4 Biomarcadores y Estudios de Gabinete
Algunos estudios de laboratorio y gabinete para auxiliar al diagnóstico de ECJ no son muy comunes; es por ello que, en el caso de países como México, por lo regular la mayor cantidad de casos son clasificados como Posible ECJ.
El electroencefalograma (EEG) es el estudio más económico que se puede aplicar, por lo regular se busca la detección de complejos periódicos de ondas agudas a una frecuencia de 1 Hz en pacientes con ECJ esporádico. El EEG representa una buena herramienta para el seguimiento de la enfermedad, sin embargo, también se han encontrado otras anomalías las cuales son detectables igualmente en Alzheimer o en Demencia por Cuerpos de Lewy (DCL) [4]. En Imagen por Resonancia Magnética (MRI) el patrón común de presentación consiste en hiperintensidades en T2 para ECJ esporádico; las anomalías resultantes en esta herramienta han demostrado ser un fenómeno temprano, siendo detectados en pacientes con mutación del gen PRNP con hasta un año de anticipación a los primeros síntomas [4]. El uso de la Tomografía por Emisión de Positrones (PET-FDG) no es muy claro en el caso de ECJ, tiene en cambio mayor utilidad para la identificación de patrones característicos correspondientes a IFF [4].
El análisis molecular del gen PRNP ubicado en el cromosoma 20 es de las mejores herramientas que tenemos a disposición para la detección de ECJ, siendo la mutación E200K la más común a nivel mundial [11], sin embargo, el método es bastante caro comparado con los anteriores, por ello es por lo que muy pocos pacientes reportan tal análisis en los casos existentes.
Existen un par de análisis con Biomarcador de Proteínas bastante efectivos si se toman en combinación: Proteína 14-3-3 y Proteína TAU, los cuales son tomados de muestra proveniente de líquido cefalorraquídeo. Para el caso de 14-3-3, es un marcador que se eleva en casos de daño neuronal, bastante útil para el diagnóstico de ECJ, pero que también se puede encontrar elevada en casos de Alzheimer o DCL [12]. TAU ha demostrado ser un marcador de diagnóstico bastante efectivo para el diagnóstico de ECJ, denotando hasta un 90% de sensibilidad y especificidad [12], sin embargo, aún no ha sido tomada en cuenta para formar parte del abanico de posibilidades para el diagnóstico de la enfermedad.
Por último, una de las nuevas y prometedoras herramientas de diagnóstico es el RT-QuIC, cuya traducción puede entenderse como “Conversión Inducida por Temblor de Priones en Tiempo Real”, siendo este una amplificación in vitro de PrPSc [12]. Reporta una sensibilidad y especificidad de cerca del 100%, lamentablemente la disponibilidad para realizarlo a nivel hospitalario es baja, al menos para México, por todo el proceso y el nivel de bioseguridad que se debe tener.
Se realizó en primera instancia una búsqueda en la bibliografía disponible de bases de datos sobre reportes y series de casos sobre pacientes de México con ECJ, dicha revisión fue tomando como referencia el procedimiento sugerido por la metodología PRISMA. La calidad de los artículos fue evaluada a través de la escala que brinda The Joanna Briggs Institute. En vista de algunas descripciones carentes de información de tales artículos, aunado a la detección de un rango temporal carente de casos, se procedió a la búsqueda de datos disponibles sobre pacientes provenientes del Instituto Nacional de Ciencias Médicas y Nutrición Salvador Zubirán y del Instituto Nacional de Neurología y Neurocirugía, para sumarles en la base de datos final. Las estimaciones no paramétricas de análisis de supervivencia de Kaplan-Meier se realizaron a través de R, así como el análisis y tratamiento de los datos [13-27].
3.1 Regresión de Cox
La regresión de Cox busca determinar la correlación entre variables independientes durante un determinado tiempo hasta un evento, como el fallecimiento, con el objetivo de determinar su efecto sobre una determinada enfermedad. Su esquematización se determina como:
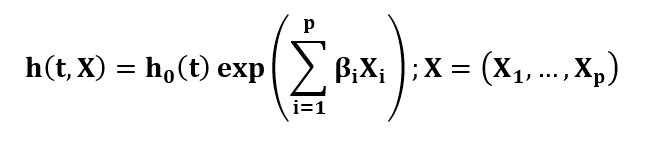
Donde h(t,X) es la función de riesgo en el tiempo t con relación a las variables predictoras, h0 (t) es la función de riesgo base en tiempo t y se toma βi como coeficiente de regresión para cuantificar el efecto de las variables efectoras (Xi), para este caso, siendo la edad βi, estratificada en 3 parámetros: 20, 50 y 100.
Fueron recopilados un total de 74 casos de México con ECJ, los cuales sus años de diagnóstico corresponden entre 1990 y 2023, se incluyeron pacientes con diagnóstico posible, probable y definitivo. Se procedió a la Estimación No Paramétrica a través de análisis de curvas de supervivencia de Kaplan-Meier con los datos recopilados de los pacientes, siendo las variables de análisis las herramientas de laboratorio y gabinete, las gráficas correspondientes se recopilan en la siguiente página web de libre acceso: https://rpubs.com/MrKristarlx07/herramientas.
Se muestra en la Figura 1 la Regresión de Cox correspondiente al banco de pacientes, siendo la variable de análisis la edad en la que se presentó el diagnóstico de la enfermedad. Una baja edad de diagnóstico de la enfermedad se asocia a una mejor supervivencia, comprobando que con pacientes de 20 años se reporta una media de supervivencia superior a un año, con lo cual se supera la supervivencia media reportada en Europa, Asia, Estados Unidos de América y Canadá [3]. Los pacientes con edad alrededor de los 50 años, en cambio, no superan el año de supervivencia, pero se acercan a los 275 días de supervivencia. Para este artículo, no se reportan los P-valores, puesto que por la naturaleza de esta patología no se cuenta con una gran población en México.
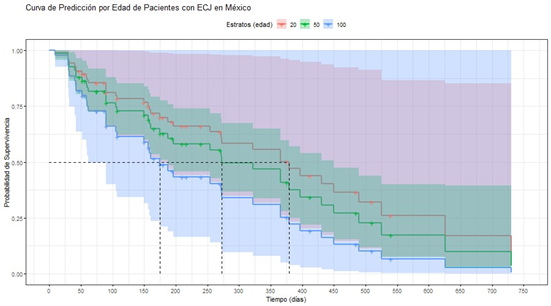
Figura 1. Curva de Predicción por Edad en Pacientes con ECJ en México
Los hallazgos principales en este artículo logran demostrar información alentadora sobre el estado de la población mexicana frente a la supervivencia en ECJ. En primer punto es importante resaltar que con nuestro trabajo se obtiene por primera ocasión el reporte de gran cantidad de casos, tanto ya publicados, como aquellos que aún no se habían visualizado en la literatura. Una característica interesante es el hecho de que, en su mayoría, en la práctica médica mexicana se realizan por lo regular EEG o RMI, lo cual, si bien nos ayuda a brindar información sobre el avance de la enfermedad, solamente nos da la posibilidad de realizar un diagnóstico de tipo “Probable”. Por otro lado, la cantidad de herramientas de laboratorio (reportadas en nuestros pacientes) utilizadas para el diagnóstico Definitivo son mínimas, generando con ello un sesgo en los pacientes, pues muchos casos Posibles y Probables con ECJ pueden en realidad ser Definitivos, y viceversa, o inclusive ser pacientes mal diagnosticados.
Se busca a futuro el análisis de la relación efectividad/coste/diagnóstico de todas las herramientas disponibles en el medio de salud mexicano. La anterior premisa puede complementarse con la revisión de la página web que se deja a disposición pública, sin embargo, es importante señalar que la aplicación o no de determinadas herramientas no influye en la supervivencia del paciente. Sin embargo, con las herramientas aplicadas actualmente, se logra rescatar que, aquellos pacientes cercanos a los 20 años reflejarán una tasa de supervivencia mayor a un año, lo cual, representa una mayor supervivencia en comparación con otras partes del mundo [3], es importante aclarar que en nuestro grupo de pacientes aquellos con menor edad tienen una tasa mayor de casos de tipo Definitivo o Probable.
La Enfermedad de Creutzfeldt-Jakob es una enfermedad rara causada por un anómalo plegamiento de la Proteína Prión, causando encefalopatías espongiformes las cuales requieren de hasta 40 años de incubación para el patógeno y tras los primeros síntomas el paciente fallecerá un año después de forma fulminante. No se tiene cura aún. Sin embargo, gracias a la aplicación de herramientas bioinformáticas se puede tener información sobre el análisis de supervivencia de los pacientes, teniendo así la construcción, en el caso de pacientes de México, una curva de predicción de supervivencia donde se clasifican por edad, siendo los casos cercanos a 20 años arrojando una media de supervivencia mayor a un año, superando así los datos epidemiológicos de supervivencia media de otras poblaciones (europea, asiática y americana del norte), lo cual puede se puede explicar investigando cuáles son los factores genéticos, epigenéticos y ambientales que caracterizan a la población mexicana, entre ellos, estando en el primer grupo anteriormente mencionado, pero aún sin estudios suficientes para su explicación: El Factor Ascendente Nativo Americano en la Población Mexicana, puesto que este factor es uno de los únicos que carece la población europea y asiática.
Para la ejecución de este trabajo, F.C.-L. fue apoyado por el Consejo de Ciencia y Tecnología del Estado de Puebla (CONCYTEP), así como por la Vicerrectoría de Investigación y Estudios de Posgrado de la Benemérita Universidad Autónoma de Puebla (VIEP-BUAP).