
Actas del Congreso Nacional de
Tecnología Aplicada a Ciencias
de la Salud

Actas del Congreso Nacional de Tecnología Aplicada a Ciencias de la Salud Vol. 6, 2024
Los Materiales Metal-Orgánicos son estructuras cristalinas porosas con diversas aplicaciones, como el almacenamiento de fármacos y catalizadores para reacciones químicas. En nuestra investigación, diseñamos un MOF basado en el 5-fluoracilo (5-flu), usado en el tratamiento de cáncer, y el Selenio 6 (6-Se), un alótropo esencial para la función celular. La combinación de 5-flu y 6-Se puede producir cristales moleculares con propiedades únicas, integrando química orgánica e inorgánica. El 5-flu facilita la formación de puentes de hidrógeno, mejorando la unión entre los cristales de 6-Se. Utilizamos Gaussian 9, para optimizar las geometrías moleculares y estudiar sus propiedades electrónicas y adsorción mediante la Teoría de Funcionales de la Densidad.
Palabras clave: DFT, métodos computacionales, química cuántica.
Metal-Organic Materials are porous crystalline structures with various applications, such as the storage of drugs and catalysts for chemical reactions. In our research, we designed a MOF based on 5-fluoracil (5-flu), used in cancer treatment, and Selenium 6 (6-Se), an allotrope essential for cellular function. The combination of 5-flu and 6-Se can produce molecular crystals with unique properties, integrating organic and inorganic chemistry. 5-Flu facilitates the formation of hydrogen bonds, improving the bond between 6-Se crystals. We use Gaussian 9 to optimize the molecular geometries and study their electronic properties and adsorption through Density Functional Theory.
Keywords: DFT, computational method, quantum chemestry
La necesidad de encontrar materiales que pudiesen ser modificados en su topología geometría, estructura cristalina, tamaños de poros y sus constituyentes mismos, da origen a los materiales de estructura metal-orgánica (Metal Orgánic Frameworks o MOF por sus siglas en inglés) [citar]. Son materiales híbridos a base de compuestos orgánicos e inorgánicos que están principalmente formadas de estas dos partes [1]. Los ligandos puente o enlazados (bringing linkers/ligands) son el constituyente orgánico como ácidos carboxílicos, enzimas, aniones, sales, compuestos heterocíclicos o polímeros a los que se les conoce como unidades de construcción secundaria (Sencondary Building Units ó SBU). La parte metálica puede ser cualquier ion o compuesto metálico, la cual es la unidad de construcción primaria (Primary Building Units PBU) y es atraída por los grupos funcionales, posiciones lábiles o vacantes dispuestas como puntos de red [2]. El ligando responde al ion metálico por medio de una posición lábil o vacante. Por lo que poseen propiedades únicas que son de interés para áreas como la adsorción química, absorción de gases, catálisis y liberación de fármacos [3]. En cuanto la liberación de fármacos, los MOFs son de utilidad debido a varias diversidades químicas y estructurales, una capacidad de carga excesiva y su capacidad de degradación [4]. En los diversos MOFs que existen, se encuentran los MOF de coordinación porosos (un tipo de polímero de coordinación poroso o PCP). Estos, generalmente están constituidos por un compuesto heterocíclico o un polímero y un metal de transición.
En esta investigación, se hace la propuesta de un tipo de MOF basado en un metaloide con uno de los alótropos del selenio, el 6-selenuro (6-Se) será utilizado como PBU y el 5-fluoroacilo compuesto heterocíclico aromático (5-flu), será utilizado como SBU. Estos materiales organo-metálicos, poseen un efecto sinérgico que potencia sus propiedades para el tratamiento de cáncer. El Selenio (Se) posee propiedades biocompatibles como: anticancerígeno y quimiopreventivo, además de ser uno de los oligoelementos esenciales para los procesos redox de las células y estar presente en más de 21 aminoácidos, proteínas, enzimas dependientes del Se como las Tiorredoxina reductasa T1-T3 [5]. Por su parte, el 5-flu es un agente antineoplásico y específicamente un compuesto orgánico análogo de la pirimidina (linker) que se utiliza en quimioterapia [6]. Se utiliza principalmente en el tratamiento de varios tipos de cáncer, así como pertenece a la clase de las antimetabolitos y actúa interfiriendo con la síntesis de ADN y ARN en las células, lo que inhibe su crecimiento y proliferación celular [7]. En un estudio realizado por Liu et. al. proponen un sistema nanométrico de liberación de fármacos anticariógenos a base de SeNP´s/5-flu. Demostraron la eficiencia y efecto sinérgico del sistema en estudios de citotoxicidad sobre células A375 (células cancerígenas epiteliales) en el proceso de apoptosis de estas [8]. Los MOF compuestos de nanopartículas tienen varias diversidades químicas y estructurales, una capacidad de carga excesiva y una alta degradabilidad; Por lo tanto, son beneficiosos como nanomedicinas tradicionales.
Enfocamos este estudio mediante simulación molecular por métodos AB INITIO utilizando Gaussian para realizar cálculos bajo la Teoría de Funcionales de la Densidad (Density Functional Theory ó DFT), como una nueva propuesta de metodología para la predicción de MOFs a base de 6-Se/5-flu. Diseñando y optimizando las geometrías moleculares y realizando los cálculos de los descriptores moleculares, los cuales nos proporcionan información importante sobre las energías de formación y cohesión con la finalidad de evaluar la viabilidad de la formación de 6-Se/5-flu. Así mismo, se investiga la estructura molecular de este tipo de MOFs que son capaces de transportar fármacos, sino también tiene una superficie bio-activa que al desintegrarse puede ser metabolizada por el cuerpo humano.
Para la construcción de los modelos, se consideraron el estado basal del Se6 y la 5-fluorouracilo (5-flu), como se muestra en la figura 1.

Figura 1. Construcción de modelos moleculares para simulación de los MOFs a base de 6-Se/5-flu
Las simulaciones se realizaron con GaussView 6.0.16 y Gaussian en su versión 9. En los cálculos por DFT, se usó el funcional B3LYP con la base 6-31G, validando parámetros de ángulos y longitudes de enlace. Se hicieron cálculos de optimización de geometrías moleculares y se calcularon frecuencias a 298.15 K para asegurar que las geometrías optimizadas representaran mínimos verdaderos, sin frecuencias imaginarias. Se calcularon niveles de energía de los orbitales moleculares HOMO y LUMO en el vacío.
Se construyeron los modelos del 6-Se y 5-flu con sus moléculas primarias hasta formar cúmulos de moléculas o clusters de los mismos. El modelo 1 estaba basado en una relación de 1:2, esto es, por cada molécula de 5-flu hay 2 moléculas de 6-Se. El modelo 2 es el inverso del modelo 1. Posteriormente, se colocan 2 cúmulos para probar las geometrías moleculares obtenidas para la construcción de los modelos 1 y 2 (ver la Figura 2 en la sección de resultados). Empleando la herramienta de condiciones de frontera periódicas (Periodic Boundry Conditions ó PBC) de Gaussview, se colocan en una celda monocíclica las estructuras moleculares del modelo 1 y 2. Posteriormente, se colocan 3 celdas adyacentes con parámetros de red a, b, y c para observar la estructura cristalina a lo largo de los X, Y, Z como se puede ver en la Figura 3.
Basado en el teorema de Koopmans, se calcularon descriptores cuánticos moleculares como: potencial químico (𝜇), dureza global (𝜂), índice de electrofilicidad (𝜔). Estos descriptores proporcionaron información sobre reactividad y estabilidad [ver Ecs. (1) - (6)] [9]. También se evaluaron las propiedades de quimisorción y fisisorción (physisorption o chemisorption), así como la energía de cohesión para analizar la geometría molecular y la fuerza de interacción [ver Ecs. (7) - (9)] [10].
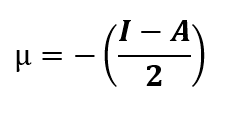
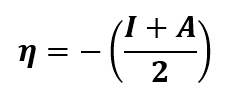
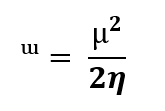
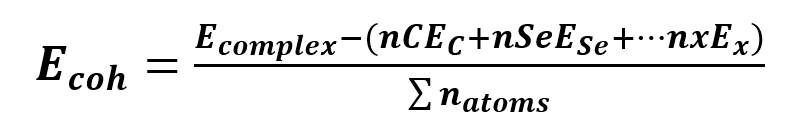
Las Figuras 2a) y 2b) muestran respectivamente, la estructura molecular de los modelos 1 y 2 de MOFs del tipo metaloide a base de 6-Se/5-flu. Esta estructura molecular presenta estabilidad química mediante los enlaces del tipo puente de hidrógeno con un átomo de Se. Las Figuras 2c) y 2d), muestran la unión de dos binas de MOFs de 6-Se/5-flu, en sus dos versiones (modelos 1 y 2), los cuales nuevamente presentan enlaces químicamente estables tanto en los enlaces puente de hidrógeno con Se (en el caso del modelo 1), como en los enlaces entre las moléculas cíclicas del 5-flo (caso del modelo 2). Además, las Figuras 3a) y 3b) muestran redes cristalinas formados por la disposición de 9 motivos dispuestos en forma adyacente de los modelos 1 y 2, respectivamente. Cada motivo que forman las redes cristalinas de los modelos 1 y 2, se forman de 2 binas de MOFs de 6-Se y 5-flu de la Figura 2. Finalmente, en la Tabla 1, se resumen los resultados numéricos obtenidos de la simulación molecular por DFT, utilizando las Ecs. (1) a (9) de la sección anterior.
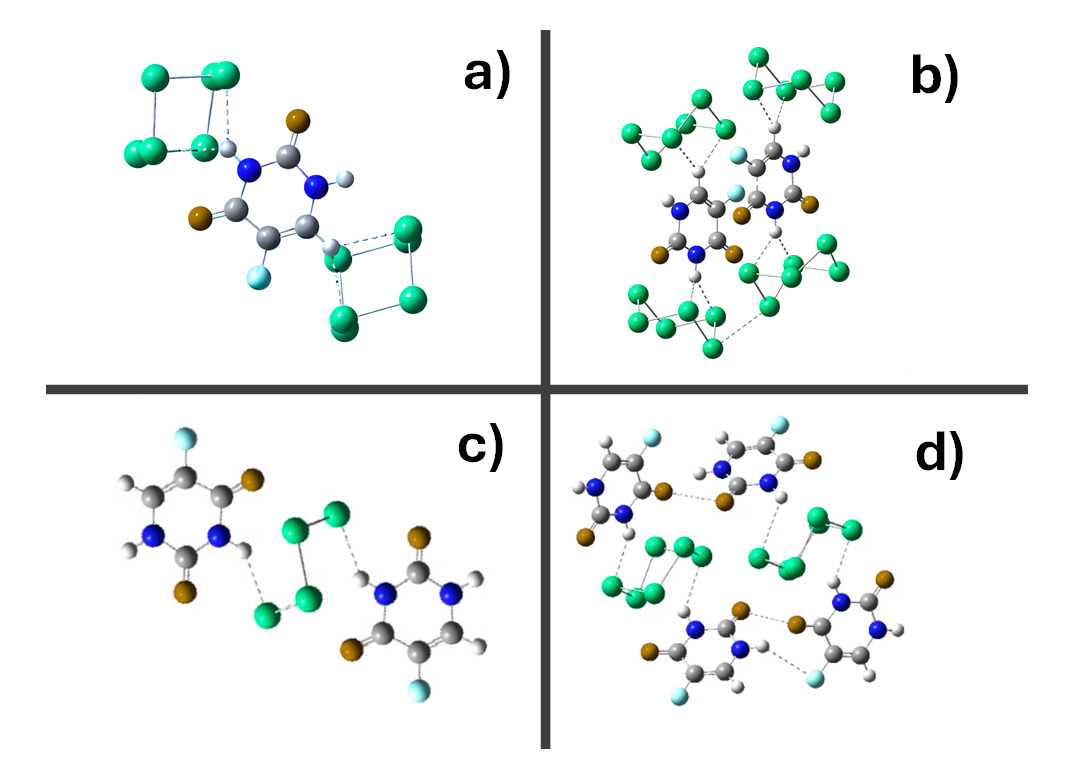
Figura 2. a) modelo 1, b) modelo 1 a, c) modelo 2, d) modelo 2a
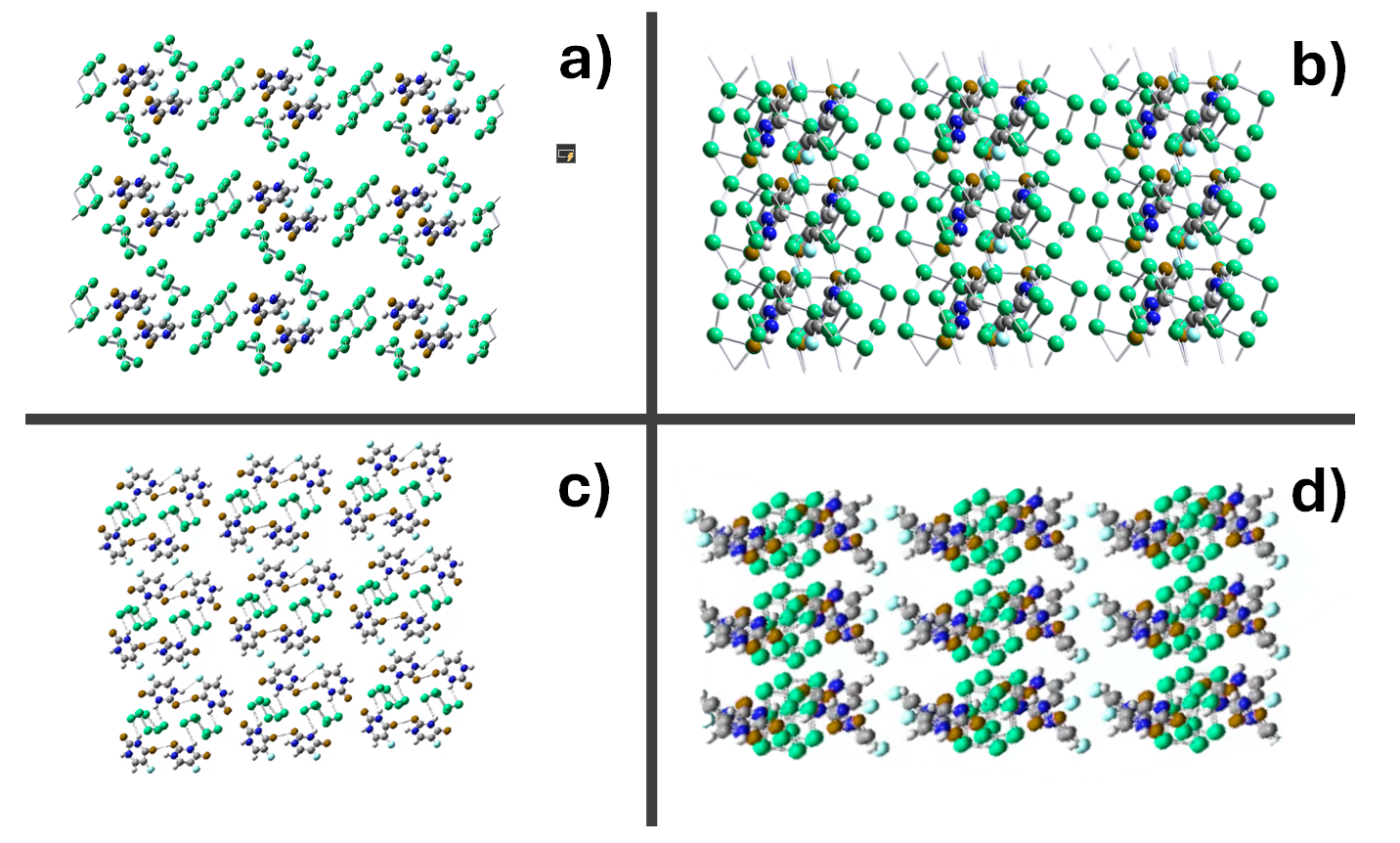
Figura 3. a) Estructura cristalina del modelo 1a con 3 celdas contiguas en a,b,c con vista en el plano (010), b) vista del plano (100) con rotación de 90°, c) Estructura cristalina del modelo 2a con 3 celdas contiguas en a,b,c con vista en el plano (010), d) vista del plano (100) con rotación de 90°
Las energías totales de los modelos 1ª y 2ª se comparan como sigue: el modelo 1a tiene una energía total mucho más negativa que el modelo 2a, lo que indica que el modelo 1a es más estable energéticamente. En las energías del HOMO y LUMO corresponden a -6.3378 eV para el Modelo 1ª, mientras que – 6.3225 eV para el Modelo 2ª. La energía del HOMO es -6.3378 eV y para el LUMO -6.3225 eV, lo que significa que ambos modelos tienen valores similares, con el modelo 1ª siendo ligeramente más estable en términos del HOMO. Asi mismo, el modelo 1ª tiene un LUMO más bajo, lo que indica que es más fácil de reducir en comparación con el modelo 2ª.
Diferencia de energía entre HOMO y LUMO, representa la estabilidad electrónica y el modelo 1ª tiene un gap más pequeño, lo que sugiere mayor reactividad en comparación con el modelo 2ª. Potencial de Ionización ambos presentan valores similares, con el modelo 1ª siendo ligeramente más difícil de ionizar. El modelo 1ª tiene una mayor afinidad electrónica, indicando que es más favorable energéticamente para agregar un electrón.
En cuanto la dureza química es la oposición de todo el cúmulo a cambios en su densidad electrónica de modo que modelo 1ª es más blando y por lo tanto más reactivo químicamente que el modelo 2ª. La electronegatividad global representa la tendencia de todo el sistema a atraer electrones el modelo 1a tiene una mayor electronegatividad, lo que indica una mayor tendencia a atraer electrones. La electrofilicidad representa la capacidad de un sistema para aceptar o repeler electrones además de describir el carácter electronegativo o electropositivo del sistema, lo que da una idea adicional para conocer el tipo de reacciones a los que será más favorable.
Tabla 1. Energía (eV) de los orbitales HOMO, LUMO, y descriptores moleculares globales de cada modelo en el vacío
| Descriptores | Modelo 1 | Modelo 2 | 1a | 2a |
|---|---|---|---|---|
| E TOTAL | -797404.075 | -419678.241 | -1594810.08 | -839357.698 |
| E HOMO | -6.6452 | -6.4828 | -6.3378 | -6.3225 |
| E LUMO | -4.0612 | -2.9937 | -4.3364 | -3.5061 |
| EGAP-MOLECULAR | 2.584 | 3.4891 | 2.0014 | 2.8163 |
| I | 6.6452 | 6.4828 | 6.3378 | 6.3225 |
| A | 4.0612 | 2.9937 | 4.3364 | 3.5061 |
| H | 1.292 | 1.74455 | 1.0007 | 1.4081 |
| µ | -5.3532 | -4.7382 | -5.3371 | -4.9143 |
| ᵚ | 11.0900 | 6.4346 | 14.2323 | 8.5753 |
| Ecoh | -120.7788 | -171.9539 | -243.4872 | -345.1241 |
| Eform | -5.0324 | -5.7317 | -5.0726 | -5.7520 |
| Eads | -393451.228 | 403780.893 | 403709.36 | -435404.851 |
El valor de la electrofilicidad para el modelo 1ª es de 14.2323 eV y el 2ª 8.5753 eV de modo que un índice de electrofilicidad positivo y más alto en el modelo 1ª, lo cual indica que este modelo es más propenso a aceptar electrones, actuando como un mejor electrófilo en comparación con el modelo 2ª. Esto puede influir significativamente en su comportamiento químico y en las reacciones en las que participa. El potencial químico es una medida de la energía potencial de una molécula para intercambiar partículas, es decir, la tendencia de una sustancia a ganar o perder electrones [11]. El modelo 1ª tiene un potencial químico más negativo que el modelo 2ª, lo que indica que el modelo 1ª tiene una mayor tendencia a atraer electrones y es más reactivo químicamente. Esto puede implicar que el modelo 1ª es más electronegativo y tiene una mayor capacidad para estabilizar cargas negativas.
La energía de formación es la cantidad de energía liberada o absorbida durante la formación de una sustancia a partir de sus elementos constituyentes en su estado estándar. Un valor negativo para la energía de formación indica que: la formación del compuesto es exotérmica, es decir, libera energía bajo estímulos externos (cambios de temperatura o introducción de energía mediante otro campo) y el compuesto es termodinámicamente estable. Un valor positivo para la energía de formación indica que la formación del compuesto es endotérmica, es decir, requiere energía, y el compuesto es menos estable termodinámicamente. Ambos modelos tienen energías de formación negativas, lo que significa que son termodinámicamente estables. El modelo 2ª tiene una energía de formación más negativa, lo que sugiere que es ligeramente más estable que el modelo 1ª. Una implicación del comportamiento del MOF de 6-Se/5-flu con morfología molecular del modelo 2ª, es como transporte y liberador de fármaco en relación 1:2 para ser un nano-medicamento viable y eficiente.
La energía de cohesión es un parámetro que evalúa la estabilidad del sistema y la fuerza con la que se mantienen unido sus constituyentes, además de ser una referencia para evaluar las geometrías moleculares. El modelo 2ª tiene una energía de cohesión más negativa, indicando que los átomos en el modelo 2ª están más fuertemente enlazados y el sólido es más estable que el modelo 1a. La energía de adsorción sirve para conocer el tipo de atracción química que existe entre las moléculas del sistema con un valor menor a 0.5 indica adsorción física, mientras que un valor mayor a 0.5 habla de una adsorción química o la formación de un enlace químico [12]. El modelo 1ª es 403709.36 eV y para el modelo 2ª es de -43404 eV. El valor obtenido para el modelo 1ª presenta adsorción química que es ideal para el tipo de proceso que nosotros evaluamos, ya que se busca en el MOFs que exista un enlace de tipo orgánico entre la parte orgánica y la inorgánica. El modelo 2ª posee un -435404.851 lo que nos habla de una adsorción física que puede deberse a las fuerzas de interacción de tipo Van der Waals, es posible que se deba al mayor número de oxígenos disponibles generando fuerzas de repulsión y atracción entre otras moléculas. Esto confirma el hecho de generar un fármaco anti-cancerígeno y bio-compatible (bio-activo y bio-degradable) utilizando el modelo 2ª del MOF de 6-Se/5-flo con relación 1:2 para la parte maloide y orgánica, respectivamente.
El modelo 1ª es más estable debido a su energía total más negativa, además, este modelo es más reactivo debido a su menor gap entre HOMO y LUMO y menor dureza molecular. En cuanto su afinidad electrónica y electronegatividad el modelo 1ª tiene una mayor afinidad electrónica y electronegatividad, indicando una mayor tendencia a atraer y ganar electrones.
Las energías de cohesión y formación del modelo 2ª tiene mayor energía de cohesión y es más fácil de formar, indicando mayor estabilidad en términos de cohesión entre átomos. De modo que modelo 1ª es más reactivo y químicamente más activo, mientras que el modelo 2ª es más estable en términos de cohesión y formación, pero más fácil de biodegradar en un medio acuoso y con pH básico. Debido a estas propiedades el MOF es altamente prometedor para el tratamiento de cáncer epitelial como sistema molecular liberador de fármacos.
Los cálculos computacionales son esenciales para el diseño y predicción de los MOFs de 6-Se/5-flo, realizarlos por medio de cálculos DFT parece ser una excelente metodología, debido a que las interacciones que hay entre moléculas orgánicas e inorgánicas en un lugar particular, puede ser completamente determinado donde se dispone la interacción específica entre átomos y grupos funcionales. Inclusive, con los valores podemos obtener información de vital importancia como su reactividad química, estabilidad y la factibilidad de la formación de los sistemas. En una extensión de este trabajo se pueden realizar cálculos para medir su área superficial especifica, porosidad y su capacidad de adsorción, así como el estudio de sus propiedades electrónicas de la cristalización de ambos modelos 1 y 2 de MOF de 6-Se/5-flo, lo cual es motivo de estudios futuros.